FREQUENTLY ASKED QUESTIONS
Below you can find answers to the most common questions about SIREs.
If you still cannot find an answer to your question, contact us!
Iron is an essential nutrient required by almost every organism. Its capacity to exchange electrons makes it essential for fundamental cell functions, like DNA synthesis, transport of oxygen and the respiratory chain. However, it is also a potential catalyst for chemical reactions involving free-radical formation and subsequent cell damage and death. Therefore, cellular iron levels have to be carefully regulated inside the cells.
Intracellular iron homeostasis is mainly regulated post-transcriptionally by the IRE/IRP regulatory system [9]. The iron regulatory proteins (IRP1 and IRP2) can recognize a cis-regulatory mRNA motif termed iron responsive element (IRE), a conserved RNA element located in the untranslated regions (UTR) of mRNAs that encode proteins involved in iron metabolism.
A canonical IRE structure is composed of a 6-nucleotide apical loop (5’-CAGWGH-3’; whereby W stands for A or U and H for A, C or U) on a stem of five paired nucleotides, a small asymmetrical bulge with an unpaired cytosine on the 5’strand of the stem, and an additional lower stem of variable length [10].
Iron regulatory proteins/iron responsive element interactions regulate the expression of the mRNAs encoding proteins for iron acquisition (transferrin receptor 1, Tfrc; divalent metal transporter 1, Dmt1), iron storage (ferritin H, Fth1; ferritin L, Ftl), iron utilization (erythroid 5-aminolevulinic acid synthase, e-Alas; mitochondrial aconitase, Aco2; Drosophila succinate dehydrogenase, Sdh), iron export (ferroportin, Fpn), oxygen availability (hypoxia-inducible factor 2 alpha, Hif2aplha) and cell cycle (CDC14A) [5,6,9]. The mRNAs of Fth1, Ftl, e-Alas, Aco2, Sdh, Fpn and Hif2alpha contain one single IRE in their 5´UTRs. The mRNA of Slc11a2 and CDC14A contain also one single IRE but in its 3´UTR and Tfrc mRNA is so far the only known mRNAs with multiple (five) IREs, all of them located in its 3´UTR (Fig. 1).

Figure 1. Human and mouse sequences of iron-responsive elements.
In response to fluctuations in the level of the labile iron pool, IRPs bind to IREs. Depending on the IRE location, they regulate gene expression by different mechanisms. Both IRPs inhibit translation initiation when bound to 5’UTR IREs, whereas their association with the 3’UTR IREs of the Tfrc mRNA mediates mRNA stabilization by preventing an endonucleolytic cleavage [11,12]. Therefore, the IRPs act as key regulators of cellular iron homoeostasis as a result of the expression control of a number of iron metabolism-related genes (Fig. 2).
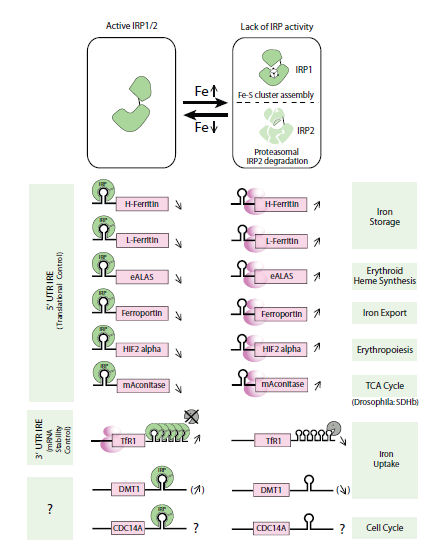
Figure 2. The iron responsive-element/iron-regulatory protein (IRE/IRP) network.[from ref. 9]
We use an in-house python program to identify IRE-like sequences in gene transcripts.
First, the sequence is screened to find the apical loop motif of the IRE corresponding to 19 different motifs and the N8 nucleotide.
Then the base pairing sequences of the upper stem and at position N7-N25 are checked allowing the presence of a single mismatch or none.
The detection of one bulge nucleotide at the right side of the upper stem or a mismatch in the upper stem or at position N7-N25 are mutually exclusive.
To ensure reliable predictions, SIREs requires a minimum sequence length of 19 nucleotides (20 for sequences with a 3' bulge, like HIF2α). While sequences as short as 19 nt can be processed, secondary structure prediction may be less accurate due to missing bases in the lower stem.
Minimum recommended sequence length: 31–32 nts:
Examples with Ftl1 IRE region:
Minimum sequence for optimal prediction and folding
TGTCTTGCTTCAACAGTGTTTGAACGGAACA (31 nt)
Minimum sequence for prediction, but suboptimal folding (NOT RECOMMENDED)
GCTTCAACAGTGTTTGAAC (20 nt)
(Missing lower stem, filled with NNNNs for folding)
Too short sequence, not detected
GCTTCAACAGTGTTTGAA (19 nt)
(Cannot find sequence match)
According to our experience we have set 3 levels of stringency (high, medium and low) for the prediction of IREs.
High and medium stringency are based on previous known and in vivo and/or in vitro well characterized IREs, i.e. the IREs present in ferritin L, ferritin H, TfR1, e-Alas, Aco2, Fpn, dSDH, CDC14A, Dmt1, Gox and Hif2aplha.
Low, stringent IRE predictions include IREs with SELEX motifs 3 to 18 with or without one single mismatch or bulge in the upper stem. Such IREs have been predicted in novel IRP-interacting mRNAs detected in a recent genome-wide study; however, although some of them have been validated in vitro, the in vivo relevance of those IREs has not yet been studied (Sanchez et al, manuscript in preparation).
To validate the predicted IREs reported by our program, we strongly recommend studying the in vitro functionality of the predicted IRE by competitive EMSA experiments as previously reported [5,6].
The minimum free energy of the predicted IRE motif calculated by RNAfold webserver is also an indicator of IRE reliability. Predicted IREs by SIREs that fail to fold with a negative free energy most probably represent non bona fide IREs.
This is highly dependent on the number of sequences and the level of detail you need in your results.
If you want to check sequences that account for less than 50000 base pairs you can use the interactive mode . It will provide you with detailed results for the predicted IREs: graphical representations of the IRES and the predicted folding, as well as as comparison with canonical validated IREs.
In case you have more than 50000 residues in your sequences, you will have to use the batch server. We limit the number of residues allowed in the Interactive Server to prevent our servers from misfunctioning due to overload. This mode will provide you with the list of predicted IREs, but not the graphical representation or the predicted folding. From there, however, you will be able to submit up to 10 sequences to the Interactive mode for a more thorough analysis.
Still, if you have a short number of sequences you may want to use the batch server, since it is about >10 times faster than the interactive one.
A new feature we introduce in SIRES 3.0 is the possibility of searching IREs by transcript ID and gene name .
If you want to predict IREs on a given transcript, you can just type its ID and easily get the prediction. We support curated NCBI transcripts (NM_ and NR_) and predicted protein-coding transcripts (XM_). We also support Ensembl transcripts
for 5 organisms with their respective prefixes: ENST (human), ENMUST (mouse), ENSRNO (rat), ENSDART (zebrafish) and FBtr (fruit fly).
If you want to screen all the curated transcripts (NM_ and NR_) and predicted protein-coding (XM_) of a given gene for IREs, without having to manually collect the sequences one by one, the gene name mode may be useful for you. You can choose both the gene and the organism before launching the search.
However, this option is a little slower than the other two, because we do internally connect to NCBI to retrieve all the sequences that match the critera. Be patient!
Also, take into account that the gene name mode does not retrieve Ensembl ids. We made this decision to speed the computation and to have more control on the results, given that the Ensembl transcript collection
for a given gene is very extensive but also contains a high proportion of non-validated transcripts, non-coding, etc.
Our interactive and batch modes allow to submit sequences in FASTA format to SIRES. However, if the format is not correct, SIREs will not be able to process the sequences.
Take the following into account:
- You need to provide an identifier with the sequence. If you only provide the sequence, it will not be detected and nothing will be computed.
- We do not accept repeated identifiers in the input. If an identifier is composed of several words separated by spaces, only the first word will be considered. Join the words with "_" if you want to keep the complete identifiers. Be careful, for example, if working with Ensemble sequences. Sequences like the ones in Fig 3. will raise an error if submitted together, given that only the transcript ID will be kept.
- If a sequence contains non-accepted characters (different from A, C, G, T, U), the program will not process it.
- Check the Troubleshooting SIREs page to get more information on possible errors and how to avoid / solve them.
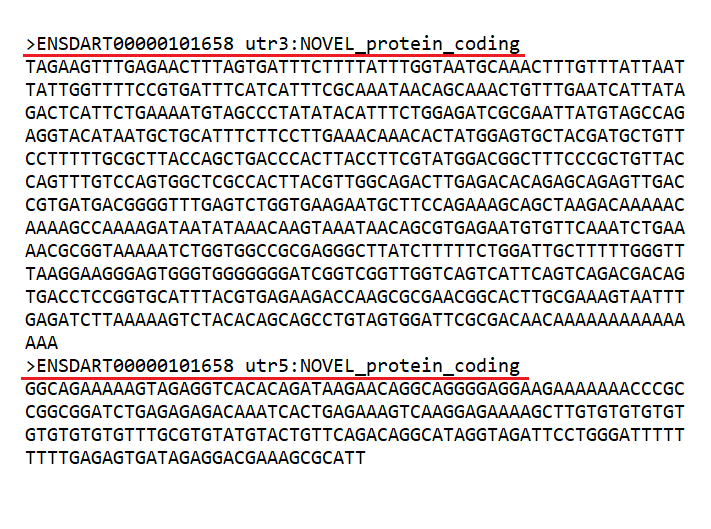
Figure 3. Example of sequences with a shared identifier.
If you made use of SIREs in your research, we would really appreciate a citation:
Campillos M, Cases I, Hentze MW, Sanchez M. Sires: Searching for iron-responsive elements. Nucleic Acids Research. 2010 Jul 1;38(Web Server). doi:10.1093/nar/gkq371