SIREs (Searching for Iron Responsive Elements) is an improved bioinformatic program that detects iron-responsive element-like motifs based on present and previous iron-responsive elements (IREs) binding studies [1, 2, 3, 4]. In its current version (3.0), SIREs is a python-based program that screens for a 19 or 20 nucleotide sequence corresponding to the core sequence of an IRE applying a series of structural constraints to produce a relevant prediction.
IREs
Structure
A canonical IRE structure is composed of a 6-nucleotide apical loop (5'-CAGWGH-3'; whereby W stands for A or U and H for A, C or U) on a stem of five paired nucleotides, a small asymmetrical bulge with an unpaired cytosine on the 5'strand of the stem, and an additional lower stem of variable length [10]. See Figure 1.
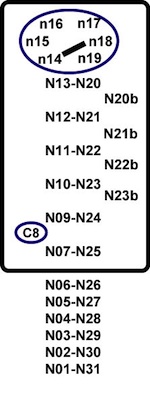
The apical loop contains a crossloop between nucleotides 14 and 18 (N14-N18) and the non-intervening nucleotides (N15 and N17) form a triloop [15].
These positions are very important for the binding to IRP1. There are more than 20 specific contacts between the IRE and IRP1, for the majority of which the C bulge (C8) and N15N16N17 (AGU) are responsible. 3' bulges are allowed in the upper stem, since these locations are not likely to take part in the interaction with the protein in complex [15].
Apical loop motifs
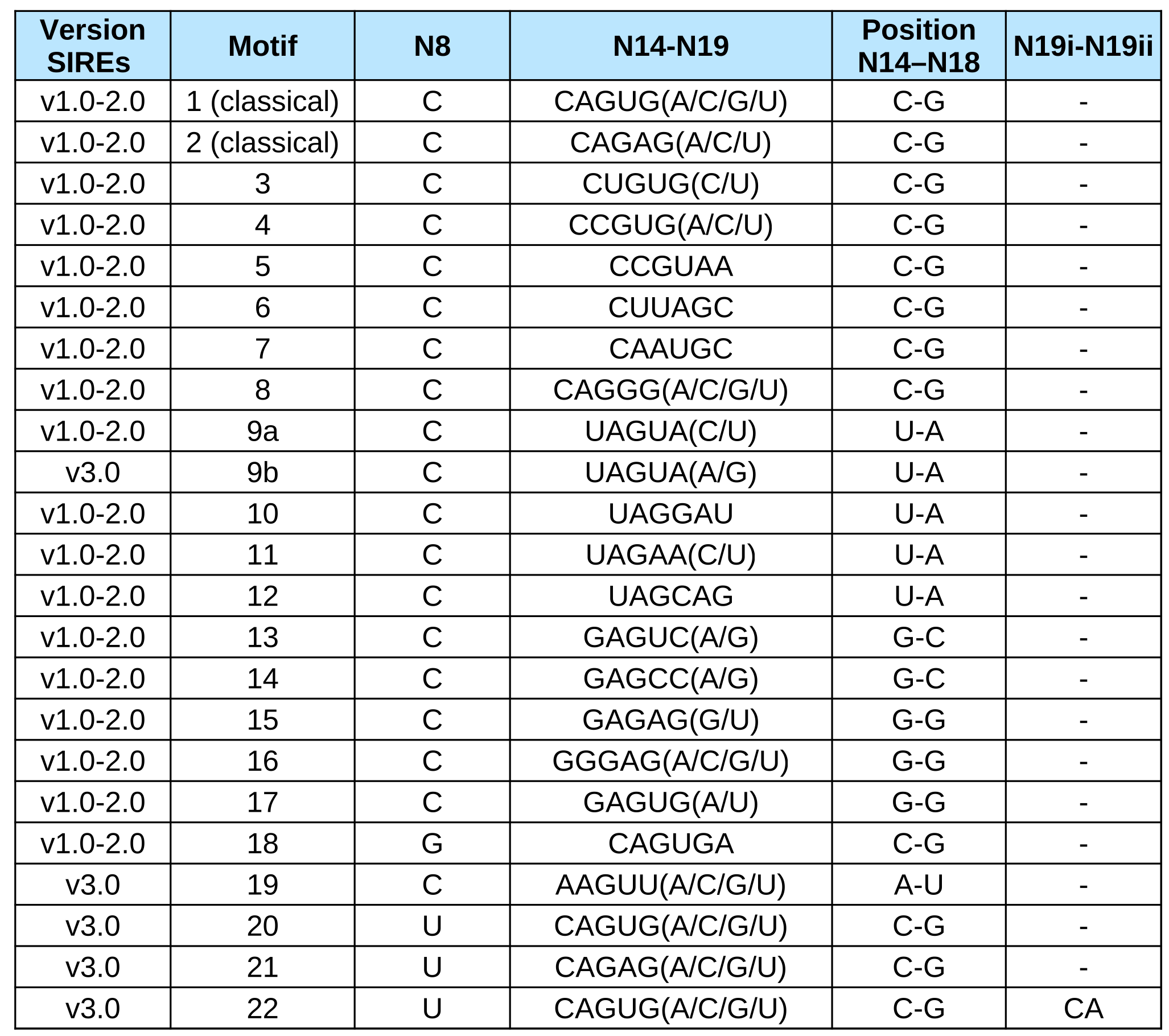
Previous SELEX (systematic evolution of ligands by exponential enrichment) experiments have reported that the 6 nucleotide apical loop of an IRE can differ from the canonical CAG(U/A)GN sequence [1, 2, 3].
Our program takes into consideration this variability and allows a total of 22 motifs (2 canonical, 19 SELEX motifs and an extra validated motif) proven to bind Iron Regulatory Protein 1 and/or Iron Regulatory Protein 2 in vitro with a relative binding efficiency bigger than 20%.
The 19 SELEX motifs differ from the canonical sequence in the sequence of the 6 nucleotide apical loop of an IRE (positions N14 to N19) or at the C bulge (N8 is a G in motif 18 and a U in motifs 20 to 22) (See Table 1). Motif 19 differs from the canonical in two positions of the apical loop (N14 is an A and N18 is an U) [16].
In Fig 1. we report the IREs that are known to bind IRPs and that have been proved to regulate iron homeostasis in vivo.
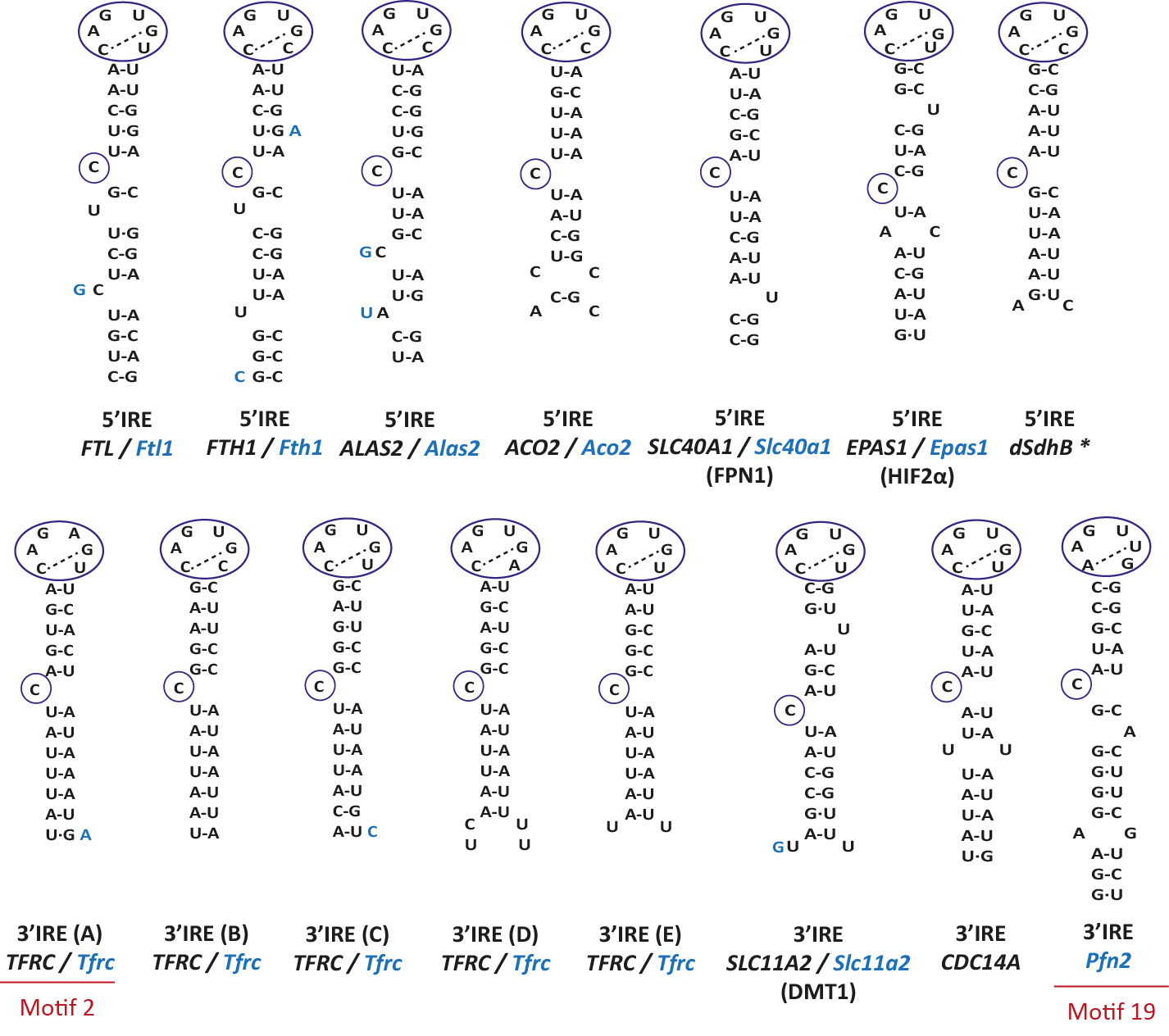
Nucleotides in blue show changes in the mouse IRE (if it exists) with respect to the human IRE.
In black, human gene; in blue, mouse gene. * dSdhB is a drosophila gene.
SIREs: algorithm
Program
SIREs is backed by an in-house Python program for sequence searching and predicts iron-responsive elements (IREs) in both RNA and DNA sequences. It assigns a confidence score to each prediction, based on sequence and structural characteristics of canonical and validated IREs.
IREs detection
Because the IREs of Dmt1 and Hif2aplha have one bulge nucleotide at the right side of the upper stem (position N21b, U, see red arrow in Figure 2), we designed the SIREs program in order to be able to detect similar type of IREs allowing one single bulge nucleotide at positions N20b, N21b, N22b or N23b. In addition, SIREs allows the detection of IRE-like motifs with a mismatch in the upper stem (positions N13-N20, N12-N21, N11-N22, N10-N23, N09-N24 or N7-N25), similarly as the one present in the Gox mRNA (see red arrow in Figure 2) [4]. The detection of one bulge nucleotide at the right side of the upper stem or a mismatch in the upper stem or at position N7-N25 is mutually exclusive.
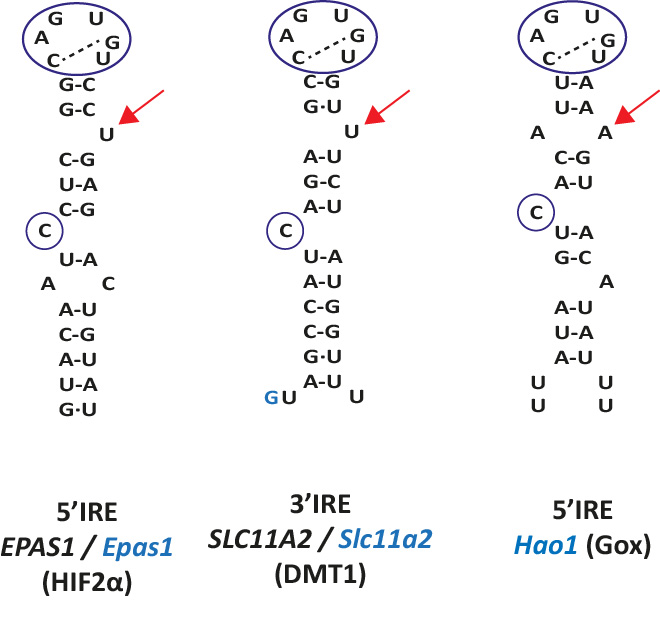
In black, human gene; in blue, mouse gene.
Recently a new motif (motif 19) was detected and validated in Pfn2 [16]. The difference between classifcal motifs and motif 19 lies in the apical loop. See red arrows in Fig 3.
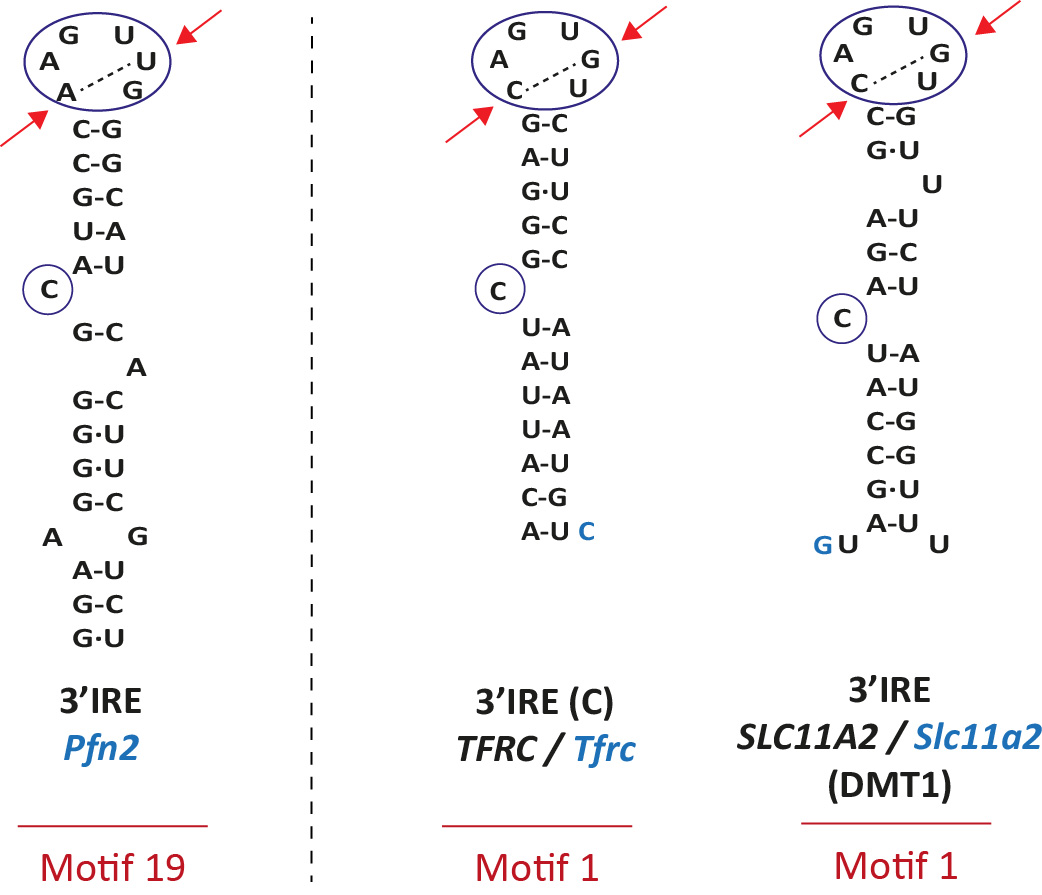
On the right, two canonical IREs (motif 1): TFRC (IRE C) and SLC11A2. In black, human gene; in blue, mouse gene.
Predictions
Predicted IRE motifs are reported as SIREs Predictions that include the 19 or 20 nucleotide sequence corresponding to positions N7 to N25. Additional 6 nucleotides from the lower stem are also reported. The RNA Folding predicted by the RNAfold Program of the Vienna Package is also reported for the predicted IRE.
Loop type
It refers to the nucleotide sequence present at positions N8 (normally the C8 bulge) and at positions N14 to N19. See Table 1.
Mismatches
It refers to the possible mismatch(es) found at positions N13-N20, N12-N21, N11-N22, N10-N23, N9-N24 or N7-N25. Base pair mismatches are reported.
Apical loop
It refers to the six nucleotides of the apical loop, positions N14, N15, N16, N17, N18 and N19.
3' Bulge
It refers to the bulge nucleotide at the right side of the upper stem (position N20b, N21b, N22b or N23b).
N25
It refers to the nucleotide at position N25. The presence of a G nucleotide at this position should be taken with caution since it may pair with the C8 nucleotide and hence impair the formation of a proper IRE.
GU/UG
It denotes the numbers of wobble base pair G.U or U.G in the upper stem or at position N7-N25. This number should be 0, 1 or 2 since we experimentally have shown that 3 or more wobble base pairs impair the formation of a proper IRE [17].
Free Energy
It denotes the minimum free energy of the predicted IRE motif calculated by RNAfold. Minimum free energy of known IREs range between -10.8 units to -3.4 Kcal/mol.
Quality
According to our experience we have modified the previous scoring system and established 6 levels of stringency for the prediction of IREs. Report features have been scored and depending on the sum value of the scores IREs are classified in the 6 categories:
| QUALITY CATEGORY | SCORES |
|---|---|
| High | [6.5, 8] |
| High-Medium | 6 |
| Medium | [4.5, 5.5] |
| Medium-Low | 4 |
| Low | [1, 3.5] |
| Very Low | <=0.5 |
High level predictions include those IREs with motif 1, 2 or 19 and either none or one single mismatch or bulge. In this category the SIREs program can detect all known
and well characterized IREs such as the ones of FTL, FTH1, TFRC, ALAS2, ACO2, SLC40A1 (Ferroportin), dSDH, CDC14A, SLC11A2 (DMT1), EPAS1 (HIF2alpha) and Pfn2. See Fig 4.
Predictions of Medium and lower qualities include IREs that do not fulfil most of the report features. To validate the predicted IREs reported by our program,
we strongly recommend studying the in vitro functionality of the predicted IRE by competitive EMSA experiments as previously reported
[5,6]

Location
It refers to the region of the transcript where the IRE is located. It is not the same as the Position,
which indicates the relative location of the IRE in the sequence provided.
The location can be: 5'UTR, 5'UTR-CDS, CDS, CDS-3'UTR or 3'UTR.
This feature is always available for Transcript and Gene Name modes, while for the Interactive and Batch modes it will only be computed if the identifier of the input sequences is a valid NCBI / ENSEMBL id.
We compute the location by mapping the relative coordinates of the predicted IRE in the input sequence to the coordinates of the features of the transcript obtained from GenBank.
Distance to cap (dCAP-5'IRE)
It refers to the distance from the middle point of the IRE sequence to the most 5' nucleotide of the sequence. Only available for 5'UTR IREs.
Distance to start codon of CDS (d5'IRE-AUG)
It refers to the distance from the middle point of the IRE sequence to the start codon of the CDS. Only available for 5'UTR IREs.
Distance to last codon of CDS (dTER-3'IRE)
It refers to the distance from the middle point of the IRE sequence to the last codon of the CDS. Only available for 3'UTR IREs.
Gold Standard score and energies
We have computed both reference energy and scores from validated IREs (motifs 1, 2 and 19) and have
included them as a visual aid in the results page. We have considered 28 IREs: 14 from human genes, 13 from mouse and 1 from drosophila. See Table 3.
The average predicted score for these IREs is: 7.36, while the average free energy of the structure is -7.11 kcal/mol.
The lowest score is the one from SLC11A2 (motif 1): 5.5.
The lowest E is from TFRC / Tfrc: -11.7 kcal/mol.
The highest E is from ACO2/Aco2: -2.8 kcal/mol.
Versions
Some major updates have been introduced in SIREs 3.0 and this may change the results obtained with previous versions of SIREs. See What is new? to learn about this.
Performance
Sensitivity, specificity and precision of SIREs 3.0 for the three different stringent levels have been calculated using a data set of 53 IRP-target genes enriched on IRE-containing mRNAs [17] and 150 random sequences that are not expected to harbor IRE elements. Values are reported in the below table.
| High | High-Medium | Medium | Medium-Low | |
|---|---|---|---|---|
| Sensitivity | 0.42 | 0.11 | 0.13 | 0.09 |
| Specificity | 0.99 | 1.00 | 0.91 | 0.98 |
| Precision | 0.92 | 1.00 | 0.35 | 0.62 |
| Balanced Accuracy | 0.70 | 0.56 | 0.71 | 0.54 |